
glp的中文全称是 “药品非临床实验管理规范 ,发展历程
药品,是人类保护自身对抗外界的重要武器结晶;在其研究过程中,细化了众多学科领域及其系统工程。因药品安全问题而产生的药害事件,历史上众多,同时也催生了众多评价体系的诞生。今天我们要聊的这个话题~GLP,即是这一过程而催生的重要评价系统,且当下已成为药品临床前重要的质量评价体系。
1、药品非临床实验管理规范(GLP)/简介
药品非临床实验管理规范(GoodLaboratory Practice,GLP),是为了保证新药临床前研究安全性试验资料的优质、真实、完整和可靠,且针对药物非临床安全性评价研究机构制定的基本要求;旨在规范新药非临床安全性研究的规范性、科学性与可重复性。
新药临床前安全性评价对新药能否进入临床研究、预测临床研究的风险程度和最终评价其开发价值起着举足轻重的作用,而一个高质量的安全性评价工作必须遵循GLP,这已是各国主管部门和新药研究单位的共识。
GLP的实施旨在规范药品安全性研究的全过程,包括试验设计、给药、观察、检测、记录、报告等,以确保试验结果能够客观、真实、全面地反映受试物的安全性特征。在缺乏科学性与规范性的基础上所产生的试验资料,将会直接影响审评员的分析、判断与综合评价,这种情况下对药品安全性进行技术审评时会存在许多问题,将会影响评价的科学性;换言之,技术审评人员只有面对规范、真实、完整的研究资料,才能在审评过程中排除诸多的干扰因素或不确定性因素,集中精力关注主要问题或更深层次问题,从而对审评对象做出更科学、合理的评价,也最终保证药品的安全、有效和质量可控性。
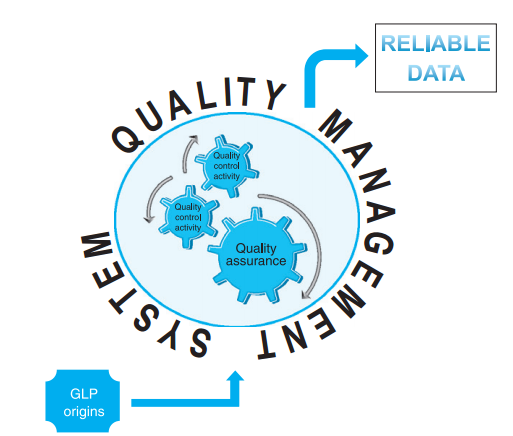
图1:GLP实验室的质量结构,图片来源于FDA官网
2、“反应停”推动GLP快速发展!
在药物毒理学发展历史上,“反应停”的悲剧无疑是促动人类对药物安全评价沉重的反思的重要事件...
1959年,西德儿科医生Weidenbaeh首先报告了一例女婴的罕见畸形,这个畸形婴儿没有臂和腿、手和脚直接连在身体上,很像海豹的肢体,故称为“海豹肢畸形儿”及“海豹胎”。医学研究表明,“海豹胎”的病因是妇女在怀孕初期服用“反应停”(沙立度胺)所致。反应停于1953年首先由西德一家制药公司合成,用于治疗早孕期间的孕吐反应有很好的止吐作用,对孕妇未见明显毒副作用,相继在51个国家获准销售。
从1956年反应停进入市场至1962年撤药,全世界30多个国家和地区共报告了“海豹胎”1万余例,各个国家畸形儿的发生率与同期反应停的销售量呈正相关,如在西德就引起至少6000例畸胎,英国出生了5500个这样的畸胎,日本约1000余例。反应停是第一个被明确为人类致畸的药物,此后全世界进行了大规模的药物致畸的研究,结果发现了不少药物有不同程度的致畸作用回。
事实上药物致畸实验并不是很复杂的实验方法,在当时的医学条件下完全可以开展这类实验,而只有美国由于官方采取了谨慎态度,没有引进这种药,因此,除自己从国外带人服用者造成数例畸胎外,基本没有发生这种病例。

图2:英国的一位海豹畸形儿和她的父亲,(图片来源:doi:10.3866/PKU.DXHX201904021)
3、美国FDA开展GLP及全球主要国家的铺开
继反应停事件后:20世纪70年代,美国FDA工作人员在审评一家大型制药公司为支持两个新药申请而提交的安全性研究报告时,发现报告中的数据前后不一致,而且有实验作弊的迹象,于是FDA对所管辖产品的安全性研究报告的可靠性产生强烈怀疑,从而对全国研究机构展开调查。调查结果显示,尽管也存在故意隐瞒对产品不利的实验结论的情况,但广泛存在于各个企业、研究机构、学校中的更严重问题是安全性实验设计、进行和报告过程中存在的缺陷,从而导致报告的可信性严重降低。
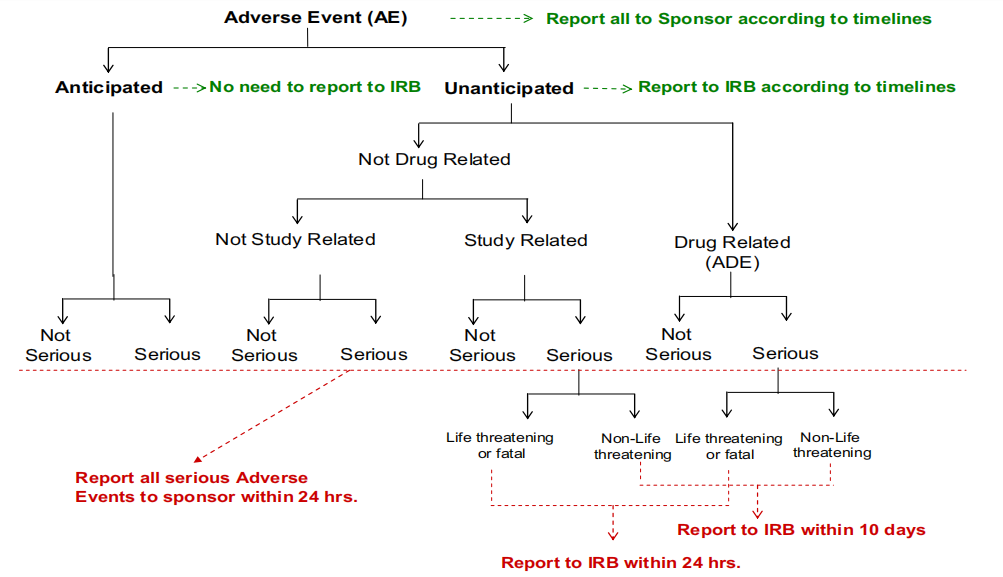
图3:FDA不良事件~主要调查员向机构审查委员会(IRB)评估报告要求,图片来源于参考资料3
针对这类情况,FDA于1976年颁布了GLP法规,规定对于此后不符合GLP标准的实验室出具的药物非临床安全性研究资料不予承认。此项法规标志着现代GLP的出现,通过强化非临床安全研究的质量管理,大大提高了药物在上临床实验前的安全性评估质量。在美国的带动下,英国、日本、法国、瑞典等国家也先后发布了本国的GLP,GLP也逐渐成为了国际上通行的确保药品非临床安全研究质量的规范。
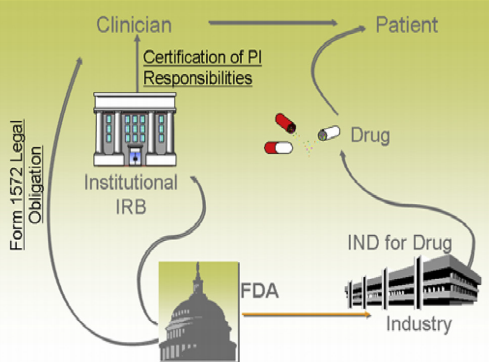
图4:FDA与临床实践,图片来源于参考资料3
在1976年美国开始试行GLP规范之后,1979年美国将其按照联邦法规正式生效;之后,1979~1980年间,欧共体制定了关于实施GLP的原则性文件。1981年,日本厚生省制定GLP规范,正式生效;并后续进行了修订和完善。1983年,欧洲国际经合组织(OECD)开始实施GLP。英、德、法、荷、意大利、瑞士、韩国等相继实施各自的GLP。
4、中国GLP的发展历程
由于建国较晚,且药品行业的特殊发展路径,我国从1991年才开始起草GLP,并于1993年原国家科委颁布,且于1994年生效。
1998年国务院机构改革后,国家食品药品监督管理局(那时候叫“SFDA”)根据国际GLP的发展和我国的实际情况,颁布了《药品非临床研究质量管理规范》,并于1999年施行。
2007年,SFDA规定未在国内上市销售的化学原料药及其制剂、生物制品,未在国内上市销售的从植物、动物、矿物等物质中提取的有效成分、有效部位及其制剂和从中药、天然药物中提取的有效成分及其制剂以及中药注射剂等的新药非临床安全性评价研究必须在经过GLP认证、符合GLP要求的实验室进行,这标志着我国从开始的GLP试行到目前强制性实施。
GLP实验室的运行成本高于同类普通实验室好几倍,GLP认证的投资也很大,约15年前的数据,建设一个完全符合GLP规范的实验室至少要5000万~6000万元,可想而知,今日再想建成一家GLP实验室,须消耗多少的投入。但即使如此,我国当前已建成符合符合国家要求的GLP实验室数十家。其数量及分布如下图所示。
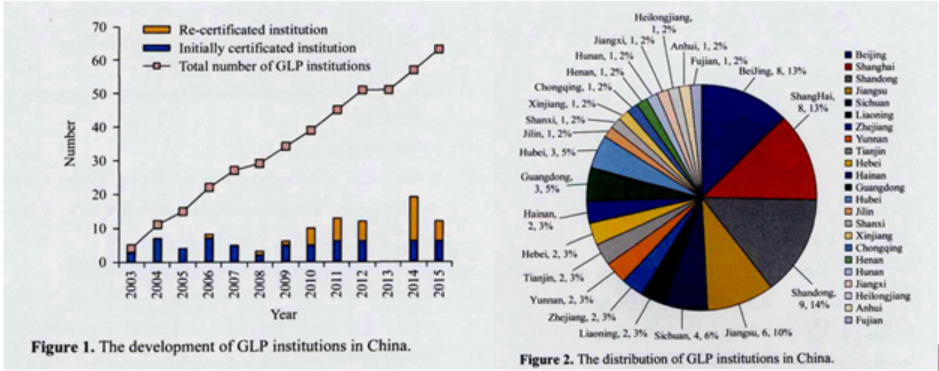
图5:国内符合要求的GLP实验室分布,图片来源于参考资料1
5、小结
我国是世界上公布实施GLP较晚的国家,与发达国家的GLP相比,差距是明显;但应该看到,经过努力,我国的GLP水平在不断提高;国内GLP的实施常规化,也助力了候选药物评价的科学性、合规性。
但在这里,笔者不得不说,GLP的合规性,绝不仅仅是文件的数量集合,一定要兼具科学性。许多所谓的GLP科研人员,在某种意义上说,仅仅是一名操作人员,且其操作往往缺少逻辑和思考。在坚持合规性的同时,往往会使人死板、教条、不思考、甚至对错误视而不见,出了问题,只会拿着自己做的账目,甩锅。GLP机构的管理,十分重要;GLP的人员,责任重大;只有真正秉持GLP的理念和要求,并有效的落实到试验评价当中,才会真正维护好GLP的初心,才能真正对得起新药发现过程中的汗水!
